









By Shang Tsai
Automation of protein sample preparation workflows to be able to address clinical questions has come of age: it is both possible and realistic, despite the hurdles outlined in our first article of this two-part series. Here, we discuss recent developments in reproducible high-throughput proteomics sample preparation workflows that do not require modification even when applied to highly disparate sample types. These advances bring proteomics a significant step closer to clinical reality.
Automated sample preparation workflows for proteomics must be simple enough that extensive training is no longer required, as it is for manual sample prep.
Not another ordinary attempt at “simplifying proteomics”
We previously identified four major bottlenecks to being able to bring proteomics techniques to the clinic: the ability to analyze enough samples (100s-1000s to more), with a simplified workflow that is both time- and cost-efficient. Below, we will describe how these new technological developments differ to previous claims and products and support the bold assertion that they truly bring proteomics in clinical settings closer to reality. Whilst tests developed with such workflows might require FDA regulatory approval, many CLIA LDTs (laboratory derived tests) that can use or could have used the instruments and workflows described herein are already on the market. We can thus anticipate that robust automation will ease and even promote the development of many new LDTs.
Automation 101 with solid phase sample prep
Automated protein sample preparation must faithfully recapitulate the underlying proteome regardless of the sample type. Further, interfering substances incompatible with downstream processing like viral transport media (VTM) detergents, salts, glycerol, PEG, Laemmli loading buffer and biological contaminants like bile components must be fully removed; this step likely requires a solid support for capturing proteins.
While non-solid support solutions for automation exist including bead- and liquid-based systems, they can require extensive optimization when working with complex biological samples and often have issues with changes of scale. Ultimately, these limitations add complexity, time and cost. In the end, the stability of a solid-phase support system remains unrivaled.
Filter-aided sample preparation (FASP) used to be frequently employed as a membrane support to remove SDS prior to mass spectrometry analysis.1 However, FASP protocols are notoriously long and membranes fail regularly, leading to experimental failure. Such disadvantages led to the development of new solid-phase capture strategies. In particular, widely adopted S-Trap™ technology allows the use of high concentrations of SDS in a fraction of the time of FASP with proteins captured on a true solid phase surface.
Independent research compared the yields from in-solution, FASP, and S-Trap based digestions of proteins extracted in SDS and urea-based lysis buffers.2 Label-free quantification was performed to analyze the differences in the identified proteome using each method. The results showed that while the different digestion methods were reproducibility within the method type, S-Trap outperformed FASP and in-solution digestions by yielding the most efficient digestion with the greatest number of unique protein identifications:
“S-Traps demonstrated the best overall performance, with the largest numbers of protein identifications and quantitative reproducibility.”2
Uniquely, the S-Trap gives the ability to explore the insoluble portion of the proteome, even from such tough substrates as bone or muscle, by first fully solubilizing the samples and then converting the formerly insoluble proteins into soluble peptides.
The S-Trap is commercially available from ProtiFi™. The product enables the extraction, solubilization and handling of all proteins in high concentrations of sodium dodecyl sulfate (SDS) prior to their further denaturation via reduction and alkylation, acidification (pH <1) and exposure to high concentrations of organic. This four-stage denaturation ensures complete destruction of undesired enzymatic activity, such as proteases and phosphatases, and maximizes efficiency of downstream digestions. Reduction and alkylation are performed in 5% SDS, precluding precipitation, or it can be performed on-column.
Denatured proteins are captured, concentrated, and rapidly cleaned of contaminants in the submicron pores of the S-Trap. Proteins are then digested in situ in a rapid (<1 hr), reactor-type digestion. Capture of protein, washing (SDS and contaminant removal) and protease addition requires less than 5 minutes. After a one- or two-hour digest at 47°C, peptides are eluted and ready for downstream processing. This process is outlined in Fig.1.
.png?width=1174&name=Fig%201%20Protifi%20S-Trap%20protocol%20(1).png)
(Fig.1) Summary of the S-Trap protocol for protein sample preparation
S-Trap sample processing technology is available either as spin columns, or automatable 96-well plates. Both formats enable the use of SDS in proteomics sample preparation. S-Trap technology has been used for samples as diverse as serum and dirt, and more recently has been extensively used to study SARS-CoV-2 infection in COVID-19 research. Further examples can be found on the ProtiFi website.3-5
Increasing throughput and efficiency: positive thinking
Simplifying the protein sample prep workflow requires a robust protocol that can be automated for protein capture, cleaning and digestion steps. The ideal protocol should be identical for the highly varied sample types of interest because sample preparation is the largest source of experimental variation, indeed 75% according to published studies.6
Complex protein samples can have heterogeneous and viscous consistencies, so automation with more robust positive pressure, rather than centrifugation, is preferable: vacuum at a maximum of one atmosphere may not be enough and centrifugation requires expensive and complex mechanisms. Positive pressure has been shown to be more reproducible than vacuum7 and typically operates at a higher pressure than vacuum approaches. Positive pressure therefore can more reliably process all the columns of a parallel set-up of a matrix of samples, such as in a 96-well plate with constant pressure over a specified with a defined pressure-profile.
Thus, a realistic assessment of the requirements of a simplified, automated protein sample prep workflows should include processing captured protein samples using a positive pressure unit with liquid-application capabilities for cleaning and digesting samples prior to analysis. S-Traps are available in a 96-well plate format which is compatible with multiple automated platforms, including the Tecan Resolvex® A200 positive pressure workstation.
ProtiFi S-Trap sample preparation in combination with the compact, benchtop Resolvex A200 positive pressure workstation offers affordable, accessible and high throughput automated proteomics sample preparation to laboratories for the first time. The Resolvex A200 system uses gas-based positive pressure to deliver maximum process reproducibility and uniformity across columns or wells and automates accurate liquid dispensing for up to 11 protocol solvents, including the S-Trap denaturation, washing, binding and elution buffers. This ensures efficient clean-up, digestion and elution.
The standard S-Trap proteomics protocols come preinstalled on the Resolvex A200 workstation, or users can create custom protocols optimized for unique needs, reducing processing times and enhancing analytical performance. A proof of principle study presented at HUPO (the Human Proteome Organization), using a model system of spiked human plasma, demonstrates the feasibility and accuracy of this workflow, and can be found here.
How far can S-Trap take us in the clinic?
Automation of the proteomics sample preparation will make a huge difference to the clinic in many key areas. One such area is in the mining of the millions of archival FFPE (formalin-fixed, paraffin embedded) samples held in labs around the world. S-Trap has already been successfully used in this area, and the standardized protocol includes the use of the HYPERsol protocol8, which has been extended to the world’s only simultaneous 96-well megasonicator, the PIXUL, as described in this HYPERsol poster.
"By processing FFPE samples with S-Trap sample processing, HYPERsol will be suitable for the automated, high-throughput analysis of clinical specimens. We thus anticipate that the HYPERsol workflow will enable novel discoveries from rich clinically annotated and histologically characterized FFPE biorepositories worldwide, thereby helping to usher in a new era of clinical proteomics."8
The HYPERsol protocol affords the best correlation ever published between paired FFPE and flash frozen samples as determined by both proteomics identifications and quantifications. Further evaluations, references and testimonials on S-Trap technology in different applications can be found on the ProtiFi website.
Having solved the bottlenecks associated with sample prep, being at last able to use the same, scalable protocol from sub-microgram to multiple mg sample sizes and having obtained new and standardized -omics data with the implementation of these simplified workflows, the next challenge is what to do with those data.
Making sense of the data
Automated sample processing enables generation of large numbers of samples which brings with it a new problem: how do we make sense of all the data? The sheer quantity of data, along with the myriad of tools and approaches can and does lead to significant errors in data analysis, due to faulty assumptions and tools or analyses being incorrectly applied. Indeed, this problem will only grow with the increasing sample throughput afforded by automation.
An efficient solution to this problem might be an online cloud-based browser-accessible -omics analysis engine that translates data into biological meaning through an easy, accessible, and interactive interface. ProtiFi recently developed such software, a world first. Called SimpliFi™, it accepts quantified -omics data of all kinds as input, obligates QC of the data and then performs many statistical analyses to allow users of all skill levels to explore and visualize their data.
The interactive analyses include differentially expressed pathways, GO enrichments, cellular markers, protein-protein interaction networks and molecular-level classifications, all of which can be performed on a smart phone. Results can be shared or published simply by sending a URL. An example of the output from the SimpliFi software is given in Fig. 2. SimpliFi uses all nonparametric statistics and resampling to provide accurate results and confidence intervals, regardless of the structure of the underlying data.
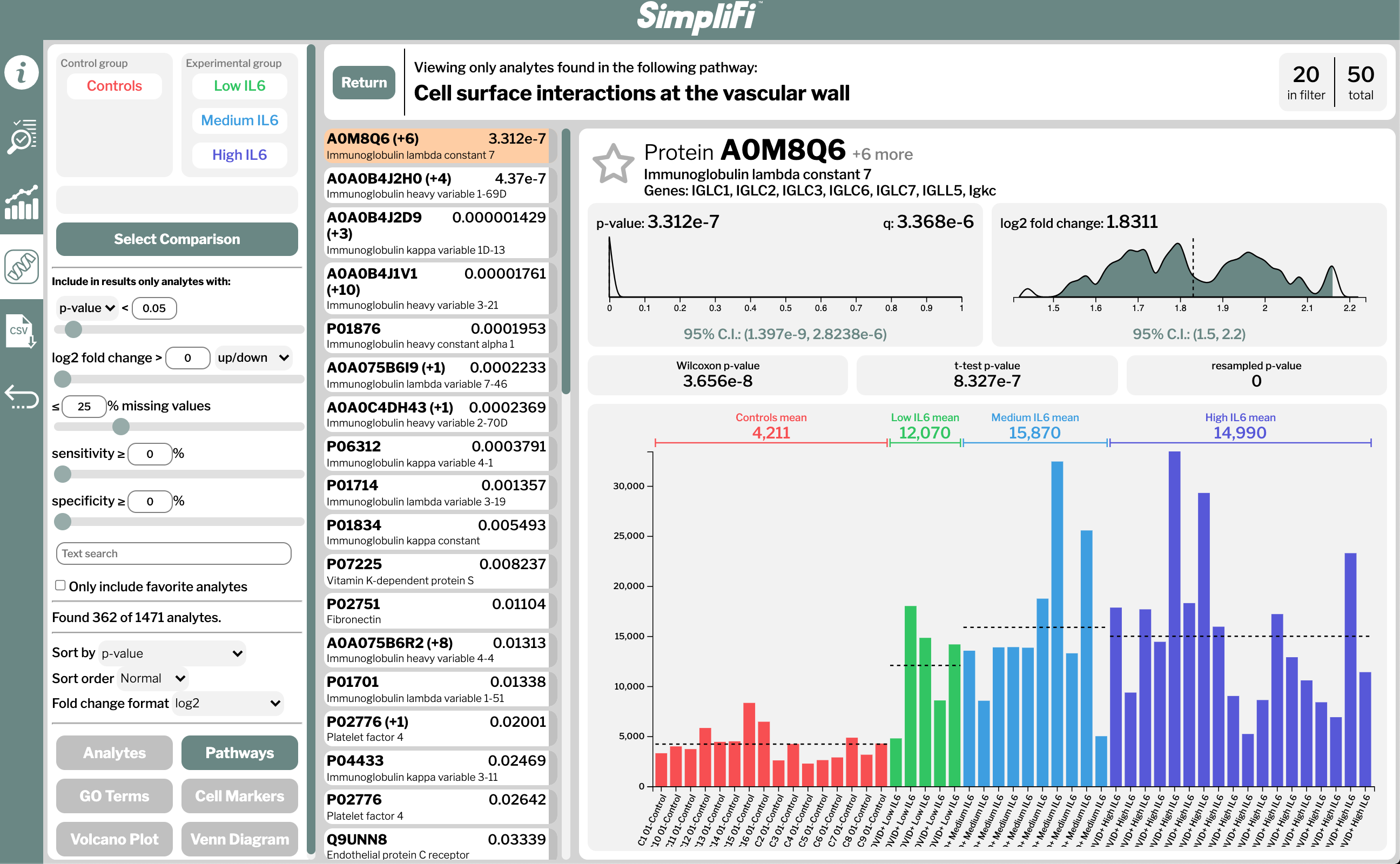
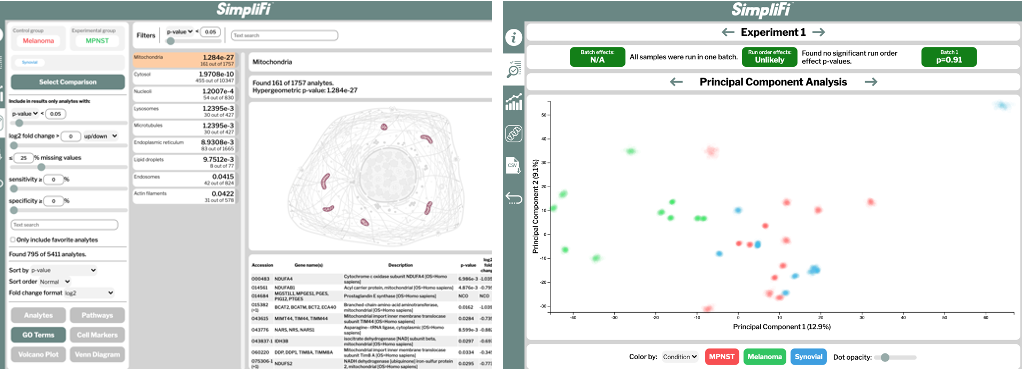
(Fig. 2) Partial output of the comparison between melanoma and control sample proteomes using ProtiFi’s “SimpliFi” software
So, can we automate protein sample prep … or not? To revisit the original set of challenges in bringing proteomics into a clinical research setting:
Can we analyze enough samples, and can we simplify our proteomics workflow, both time-efficiently, and cost effectively?
Yes, we can, because Tecan and ProtiFi have partnered to create an integrated, highly scalable, easy-to-use proteomics workflow powered by the ProtiFi S-Trap™ sample preparation system and the Tecan Resolvex A200 automated positive pressure workstation.
Can we then analyze and understand those data?
Yes, SimpliFi™ allows non-omics experts to use these powerful tools to interrogate and understand biological questions. Watch the short video here to see for yourself how proteomics could finally clinch its long overdue place pre-clinical research and then finally in clinical applications.
Reproducible Automated Proteomics and Multiomics Sample Preparation
Would you like to learn how to ensure reproducible results for your automated proteomics workflows?

References
- 1. Wiśniewski, J. R., Zougman, A., Nagaraj, N., & Mann, M. (2009). Universal sample preparation method for proteome analysis. Nature methods, 6(5), 359–362. PMID: https://pubmed.ncbi.nlm.nih.gov/19377485/ DOI: https://doi.org/10.1038/nmeth.1322
2. Ludwig, K. R., Schroll, M. M., & Hummon, A. B. (2018). Comparison of In-Solution, FASP, and S-Trap Based Digestion Methods for Bottom-Up Proteomic Studies. Journal of proteome research, 17(7), 2480–2490. PMID: https://pubmed.ncbi.nlm.nih.gov/29754492/ DOI: https://doi.org/10.1021/acs.jproteome.8b00235
3. D'Alessandro, A., Thomas, T., Dzieciatkowska, M., Hill, R. C., Francis, R. O., Hudson, K. E., Zimring, J. C., Hod, E. A., Spitalnik, S. L., & Hansen, K. C. (2020). Serum Proteomics in COVID-19 Patients: Altered Coagulation and Complement Status as a Function of IL-6 Level. Journal of proteome research, 19(11), 4417–4427.PMID: https://pubmed.ncbi.nlm.nih.gov/32786691/ DOI: https://doi.org/10.1021/acs.jproteome.0c00365
Click here to explore this dataset with SimpliFi software.
4. Thomas, T., Stefanoni, D., Dzieciatkowska, M., Issaian, A., Nemkov, T., Hill, R. C., Francis, R. O., Hudson, K. E., Buehler, P. W., Zimring, J. C., Hod, E. A., Hansen, K. C., Spitalnik, S. L., & D'Alessandro, A. (2020). Evidence of Structural Protein Damage and Membrane Lipid Remodeling in Red Blood Cells from COVID-19 Patients. Journal of proteome research, 19(11), 4455–4469.PMID: https://pubmed.ncbi.nlm.nih.gov/33103907/DOI: https://doi.org/10.1021/acs.jproteome.0c00606
Click here to explore this dataset with SimpliFi software.
5. Appelberg, S., Gupta, S., Svensson Akusjärvi, S., Ambikan, A. T., Mikaeloff, F., Saccon, E., Végvári, Á., Benfeitas, R., Sperk, M., Ståhlberg, M., Krishnan, S., Singh, K., Penninger, J. M., Mirazimi, A., & Neogi, U. (2020). Dysregulation in Akt/mTOR/HIF-1 signaling identified by proteo-transcriptomics of SARS-CoV-2 infected cells. Emerging microbes & infections, 9(1), 1748–1760.PMID: https://pubmed.ncbi.nlm.nih.gov/32691695/DOI: https://doi.org/10.1080/22221751.2020.1799723
6. Piehowski, P. D., Petyuk, V. A., Orton, D. J., Xie, F., Moore, R. J., Ramirez-Restrepo, M., Engel, A., Lieberman, A. P., Albin, R. L., Camp, D. G., Smith, R. D., & Myers, A. J. (2013). Sources of technical variability in quantitative LC-MS proteomics: human brain tissue sample analysis. Journal of proteome research, 12(5), 2128–2137.PMID: https://pubmed.ncbi.nlm.nih.gov/23495885/DOI: https://doi.org/10.1021/pr301146m
7. https://www.americanlaboratory.com/914-Application-Notes/172423-Evaluation-of-an-Automated-Solid-Phase-Extraction-Method-Using-Positive-Pressure/ Accessed 22 March 2020
8. Dylan M. Marchione, Ilyana Ilieva, Kyle Devins, Danielle Sharpe, Darryl J. Pappin, Benjamin A. Garcia, John P. Wilson, John B. Wojcik. HYPERsol: High-Quality Data from Archival FFPE Tissue for Clinical Proteomics. Journal of Proteome Research 2020, 19 (2) , 973-983. PMID: https://pubmed.ncbi.nlm.nih.gov/31935107/ DOI: https://doi.org/10.1021/acs.jproteome.9b00686
Keywords:
About the author

Shang Tsai
Shang Tsai is the Head of Marketing and Product management for Tecan SP. He and his team support and work with customers globally to develop innovative sample preparation solutions and efficient workflow for their analytical needs. Shang has over 25 years of analytical science experience with roles within marketing, business development, applications, and product development.