









By Beatrice Marg-Haufe
Written in collaboration with Zymo Research, Irvine, CA, USA.
With the advent of next-generation sequencing (NGS), the field of metagenomics has exploded in recent years, as scientists are now able to study microbes as communities instead of individual organisms. This has revolutionized our understanding of the relationships between microbiota, human health, and the environment.
 Getting a true picture of the microbiome requires uprooting bias inherent in sample preparation.
Getting a true picture of the microbiome requires uprooting bias inherent in sample preparation.
Whilst the boom in metagenomic analysis of the microbiome has resulted in many new discoveries, data reproducibility has remained a challenge. The issue of reproducibility spans metagenomic research across labs, and stems from the fact that bias can be introduced at various steps along the metagenomics workflow, as observed by many in the field [1-6]. The problem of bias is so widespread that even submitting the same sample to two different microbiome profiling organizations can yield results that are dramatically different from one another [7].
These biases can arise at every step throughout the entire metagenomics workflow. However, one of the biggest contributors to bias lies in the nucleic acid extraction step. With growing evidence of systemic biases, the need for more representative nucleic acid extraction in the metagenomics workflow is now greater than ever.
How do biases occur within extraction?
Microbial communities are complex and diverse, consisting of Gram-positive bacteria, Gram-negative bacteria, and fungi. Accurate metagenome profiling requires liberation of DNA from all the diverse species within a microbial community. However, it is common to observe ineffective lysis during the nucleic acid extraction, which then leads to microbial profile bias. This is because some microbes are very difficult to lyse [6, 8]. If the cells are not lysed, the DNA will remain locked away within the cell, and will not be purified or detected.
It has been shown that processes utilizing chemical or thermal lysis result in an over-representation of the easy-to-lyse organisms (Gram-negative bacteria). Since the tough-to-lyse organisms (e.g. Gram-positive bacteria and yeast) are more resistant to DNA liberation, this causes a bias towards the easy-to-lyse species. Many extraction protocols do not account for these vast differences in sample composition, so it is common to observe non-uniform lysis and subsequent microbial profile bias [9].
Extraction protocols that utilize mechanical lysis (e.g. sonication, blending, liquid nitrogen/mortar and pestle, French pressing, and bead beating) are considered the best approach to microbial lysis due to their stochastic nature, with bead beating referred to as the gold standard[4]. However, these mechanical lysis methods still need to be optimized or they will suffer from issues such as low yield, excessive nucleic acid shearing, non-uniform lysis, excessive heat, and shear forces.
Bias-free methods
The ZymoBIOMICS® line addresses this key challenge of bias within a metagenomics workflow. The ZymoBIOMICS 96 DNA Magbead Kit utilizes mechanical lysis that has been developed and optimized with microbial community standards to ensure complete lysis of all the tough-to-lyse organisms (Figure 1).
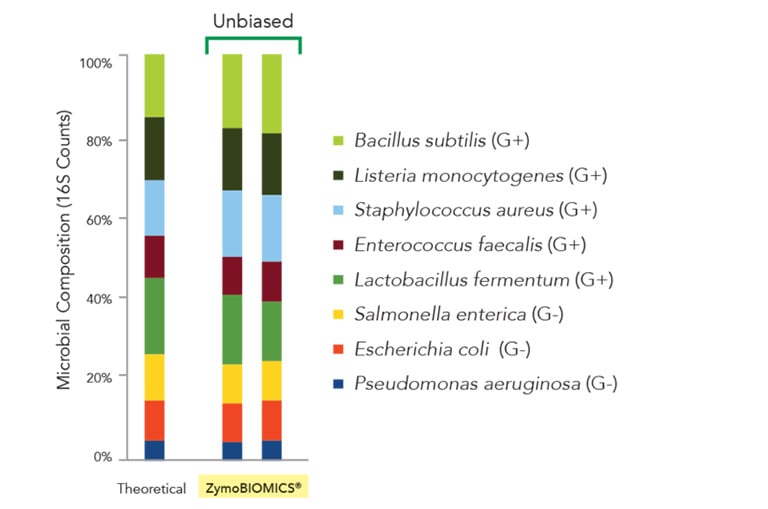
Figure 1: The ZymoBIOMICS Microbial Community Standard was purified using the ZymoBIOMICS-96 DNA Magbead Kit to evaluate the level of bias introduced from the purification. The eluted DNA was subjected to 16S rRNA gene targeted sequencing with an internal library preparation protocol. The microbial composition was determined by mapping raw sequencing reads against reference 16S sequences of the strains contained in the standard. The composition of two individual extractions of the ZymoBIOMICS microbial standard was compared to the theoretical composition of the standard and shown to match closely, indicating unbiased lysis.
Now we’ve seen how bias can arise in metagenomic analysis, and how to prevent it, we need to ensure that this lack of bias is maintained when the DNA is used in downstream applications, such as library preparation ahead of NGS. Ideally the extracted microbiome DNA is sufficiently pure and of high enough quality to be used directly, without further purification.
Find out how to optimize and streamline this process in the next article of our blog series.

Acknowledgements
This blog was written in collaboration with Zymo Research, Irvine, CA, USA.
References
- Sinha R, Abnet CC, White O, Knight R, Huttenhower C: The microbiome quality control project: baseline study design and future directions. Genome Biol 2015, 16:276.
- Hsieh YH, Peterson CM, Raggio A, Keenan MJ, Martin RJ, Ravussin E, Marco ML: Impact of Different Fecal Processing Methods on Assessments of Bacterial Diversity in the Human Intestine. Frontiers in microbiology 2016, 7:1643. 13.
- Vishnivetskaya TA, Layton AC, Lau MC, Chauhan A, Cheng KR, Meyers AJ, Murphy JR, Rogers AW, Saarunya GS, Williams DE et al: Commercial DNA extraction kits impact observed microbial community composition in permafrost samples. FEMS microbiology ecology 2014, 87(1):217-230. 14.
- Hart ML, Meyer A, Johnson PJ, Ericsson AC: Comparative Evaluation of DNA Extraction Methods from Feces of Multiple Host Species for Downstream Next-Generation Sequencing. PloS one 2015, 10(11):e0143334. 15.
- Kennedy NA, Walker AW, Berry SH, Duncan SH, Farquarson FM, Louis P, Thomson JM, Satsangi J, Flint HJ, Parkhill J et al: The impact of different DNA extraction kits and laboratories upon the assessment of human gut microbiota composition by 16S rRNA gene sequencing. PloS one 2014, 9(2):e88982. 16.
- Sohrabi M, Nair RG, Samaranayake LP, Zhang L, Zulfiker AH, Ahmetagic A, Good D, Wei MQ: The yield and quality of cellular and bacterial DNA extracts from human oral rinse samples are variably affected by the cell lysis methodology. Journal of microbiological methods 2016, 122:64-72.
- Saey TH: Here is the poop on getting your gut microbiome analyzed. In: Science News. vol. 2017; 2014.
- Farkaš V, Takeo K, Maceková D, Ohkusu M, Yoshida S, Sipiczki M. Secondary cell wall formation in Cryptococcus neoformans as a rescue mechanism against acid-induced autolysis. FEMS Yeast Research, 2009, 9(2): 311-320.
- Costea et al. Towards standards for human fecal sample processing in metagenomic studies. Nature Biotechnology (2017) 11:1069-1076.
About the author

Dr Beatrice Marg-Haufe
Dr. Beatrice Marg-Haufe is a product manager at Tecan Switzerland with over 10 years of experience in assay development and product management. She studied biochemistry at the University of Bielefeld, Germany, and at Harvard Medical School, USA. She focused on cancer research during her PhD in Biochemistry at the MPI, Munich, Germany. She joined Tecan in 2009 focusing on applications for the agriculture and genomics market.