









By Laura Nea
Is your business IVDR-ready, or are there treacherous gaps in your strategy? This November marks the halfway point in the five-year transition to the In Vitro Diagnostic Regulation (IVDR) 2017/746—a major regulatory overhaul that calls for reclassification and recertification of all IVD devices registered in the European Union. With its expanded scope and more stringent requirements, IVDR impacts the entire supply chain. The May 2022 transition deadline may seem a long way off, but there’s no time to lose. In this 2-part series, we help you take stock of the situation, with a special focus on how to prepare when it comes to managing OEM relationships and new partnerships.

Marketing IVD devices in Europe: Is your business IVDR-ready?
IVDR: blockbuster or showstopper?
The definition of an in vitro diagnostic (IVD) medical device encompasses a wide range of laboratory developed tests, point-of-care devices, instruments, reagents and kits used to analyze human samples and guide clinical decision making.
Until now, most governments have taken a relatively lenient stance on regulation of IVD devices compared to pharmaceutical drugs and medical devices that come into direct contact with patients. The In Vitro Diagnostic Regulation (IVDR 2017/746) brings European regulations for IVD devices into closer alignment with the more rigorous requirements for other medical devices, and seeks to make the regulatory process more “robust, transparent, predictable and sustainable.”
Some of the intended benefits of IVDR include:
- Higher standards of quality and safety for IVD devices
- More clearly defined definitions and regulatory scope
- Improved oversight and traceability across the supply chain and throughout the device lifecycle
- Better ability to accommodate new technologies and scientific advances
- Facilitation of global trade through the promotion of more convergent regulations
While the IVDR should ultimately be a positive step forward for the industry, most IVD manufacturers still have a lot of work ahead of them to ensure a smooth transition and avoid IVDR becoming a “showstopper” that freezes them out of EU markets.
Timeline to the new IVD regulation
The IVDR came into force in May 2017, setting the clock ticking on the five-year transition period for manufacturers selling IVD medical devices into European markets (Fig 1).
As the IVDR is phased in, the current IVD Directive (IVDD 98/79/EC) will be phased out. With no grandfathering of devices already on the market, all IVDs must be transitioned to the new regulation.
While five years may seem like ample time to make the transition, we are already at the halfway mark, and for most businesses there is still much more to be done and considerable uncertainty in the process, particularly since notified bodies (NB) are going through the transition process themselves, and the first notified body designation under IVDR has only just been announced¹.
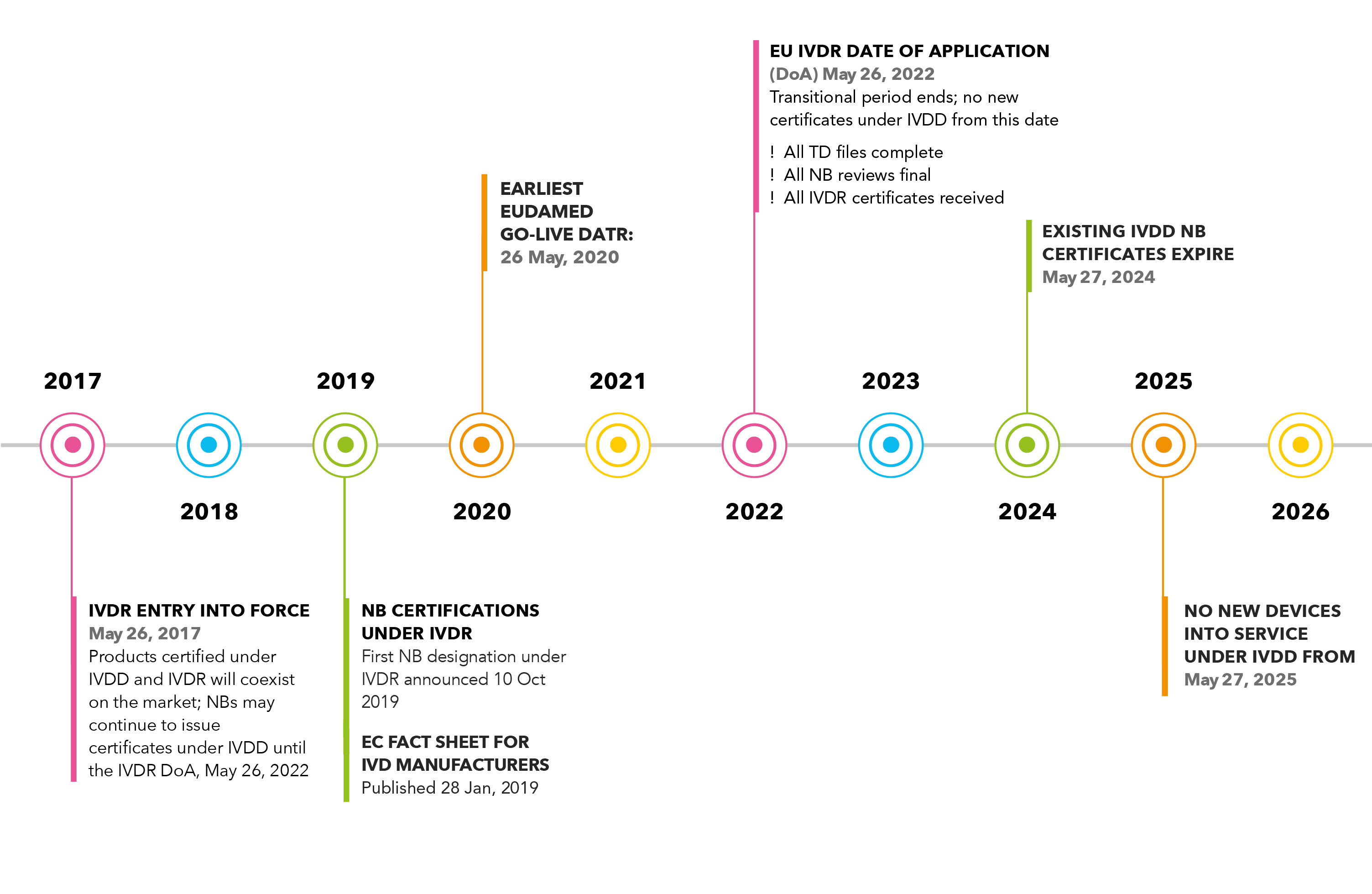 Figure 1. Timeline to IVDR 2017/746 (Click to view larger). The database of NB certifications (NANDO) is found here. The EU Fact Sheet for IVD Manufacturers also contains more information about the timeline.
Figure 1. Timeline to IVDR 2017/746 (Click to view larger). The database of NB certifications (NANDO) is found here. The EU Fact Sheet for IVD Manufacturers also contains more information about the timeline.
IVDR: Five challenges of the change
Before we consider what you can do at this point to ensure as smooth a transition as possible, here is a brief recap of some of the most notable changes and challenges the IVDR brings:
1. Extended IVD definition and scope.
Under IVDR, the definition of an IVD has been clarified and now encompasses more types of devices, including relatively new technologies like predictive genetic tests, companion diagnostics and even software. The new regulation also makes it clear that regulatory oversight extends to devices such as home-use genetic tests that are made available to EU citizens through “distance sales” (e.g. over the internet) by companies based outside the EU—even if the tests themselves are carried out in non-EU countries.
2. New device classification system.
One of the most dramatic changes introduced by the IVDR is the replacement of the current list-based classification system with a clear set of rules for assigning each device to one of four risk categories, ranging from lowest risk (class A) to highest (class D). Under the IVDD classification system, notified body (NB) oversight was only required for products expressly listed in Annex II of the directive (about 20% of IVDs); the vast majority of IVD devices could “self-certify” without any need for any NB involvement. The new regulation turns this ratio on its head, with about 80% of devices expected to fall into risk classes B, C or D, all of which require some level of NB oversight. Clearly this greatly increases the regulatory burden for IVD manufacturers and their NBs.
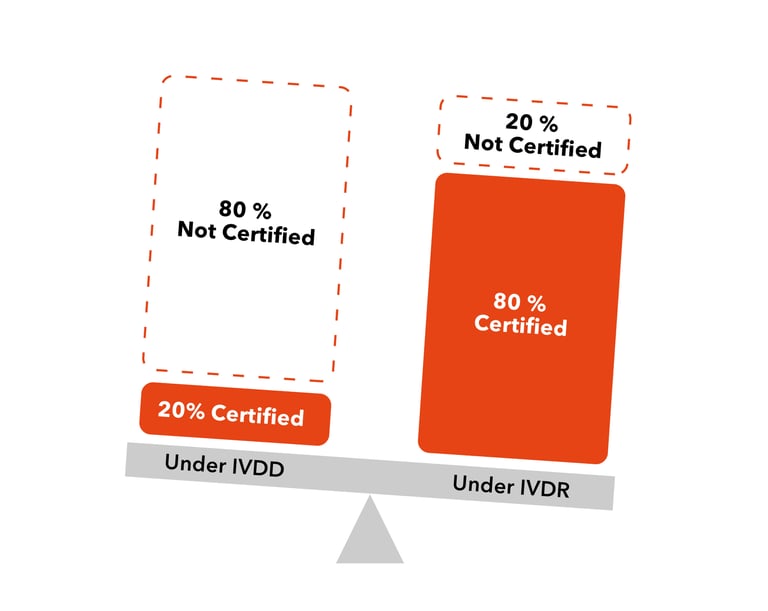
Figure 2. IVDR requires many more IVD devices to be NB-certified
3. Likely NB bottlenecks.
With tighter regulations and more IVDs requiring oversight, the workload for NBs will rise sharply. At the same time, the number of available NBs is shrinking. While 11 NBs under IVDR are reported to have submitted applications for certification, only one has been announced to date1. When IVDR-designated NBs do come online, they will likely prioritize working with companies who have already established a relationship with them in advance and have all their technical documentation in order.
4. Lifecycle approach.
Compared to IVDD, IVDR takes a more holistic approach that spans the entire product lifecycle, with more comprehensive measures to encourage continuous evaluation both pre- and post-launch. IVDR retains all existing IVDD requirements and adds new ones, including:
- Requirements for manufacturers to establish and demonstrate effective quality management systems (QMS)
- More stringent requirements for clinical evidence demonstrating conformity
- New requirements for post-market performance monitoring and reporting
- Introduction of unique device identifiers (UDIs) for improved traceability throughout the device lifecycle
5. Greater supply chain oversight and device traceability.
The new regulations have been extended to cover the entire supply chain, adding additional responsibilities for manufacturers as well as other economic operators - importers, distributors and authorized representatives. Each link in the supply chain has responsibility to check the previous link, and legal manufacturers must appoint a Person Responsible for Regulatory Compliance (PRRC) to ensure IVDR compliance across the chain. IVDR also gives NBs discretionary authority to audit suppliers and subcontractors if needed.
Keep calm, but don’t just “carry on”
As we approach the halfway mark in the IVDD-IVDR transition, now is a good time to sanity-check your progress and close any gaps in your transition strategy. Here are a few important items for your checklist:
Infrastructure: Is your current business model working for you when it comes to executing your transition plan? Are C-suite leaders including CFO and COO appropriately engaged, and is there a dedicated team empowered to drive the strategy forward?
Reclassification: Have all IVD products in your pipeline and on the market been classified according to the new rules? Has a tracker been set up to indicate certification strategy (renew IVDD certificate or pursue IVDR certification) and NB submission timeline for each device?
Portfolio review: Have cost-benefit and ROI studies been carried out? Does it make sense to rationalize your portfolio or make adjustments to the product pipeline? Does your strategy include exploration of non-EU markets and collaborations outside the IVD medical device industry?
NB alignment: Have reclassified products been mapped against NB requirements, and suitable NBs identified? Have you reached out to NBs to enquire whether they have applied for IVDR designation? Can they send you guidelines for their application and audit process?
Gap analysis: Assess technical documentation, clinical evidence, labelling and packaging, QMS, post-market surveillance and reporting. Have available technical and clinical data been reviewed against conformance requirements? Have plans been established to acquire outstanding support data?
QMS: Have you reviewed current QMS against the new regulations? Has your QMS been updated accordingly? Has a PRRC been appointed and appropriately trained to ensure compliance across the supply chain?
Transition budget: What is the burn rate to date, and does it match with projected spend to the end of the transition period?
Quality contracts: Have quality contracts with existing suppliers, contract manufacturers and other economic operators been reviewed and updated as needed with provisions to facilitate IVDR compliance?
What about outsourcing and OEM partnerships?
In this shifting regulatory landscape, OEM partnerships can be a source of great experience and guidance, or they can become your worst nightmare. In light of the extended scope of regulatory oversight that IVDR brings, it is more important than ever to partner with companies you trust to help you design and develop high-quality compliant IVD devices, supply you with the technical expertise and data you need for product certification, and ensure compliance can be maintained throughout the device lifecycle. And of course, those partners must have a quality management system that is compliant with IVDR requirements.
We’ll go into more detail about what to consider when working with OEM partners in Part 2 of this series, so stay tuned to The Blog for the next article.

References
1. Brennan, Z. (2019, October 10). EC Unveils First Notified Body Designation under IVDR. Retrieved from https://www.raps.org/news-and-articles/news-articles/2019/10/ec-unveils-first-notified-body-designation-under-i
About the author

Laura Nea
Laura Nea is Vice President of Global Regulatory Affairs at Tecan. Laura and the multinational Global Regulatory team partner with Tecan’s customers to ensure successful strategies for commercialization of products worldwide. Laura has over 30 years experience in the diagnostics and IVD market and has been on the Tecan team since 2010.